Comparative Analysis of Electronic Structures Calculations: A Simple Test Case Set for Kohn-Sham Density Functional Theory and Hartree-Fock Methods
DOI:
https://doi.org/10.55749/ijcs.v2i2.33Keywords:
Hartree-Fock, DFT, Electronic structures, OrbitalsAbstract
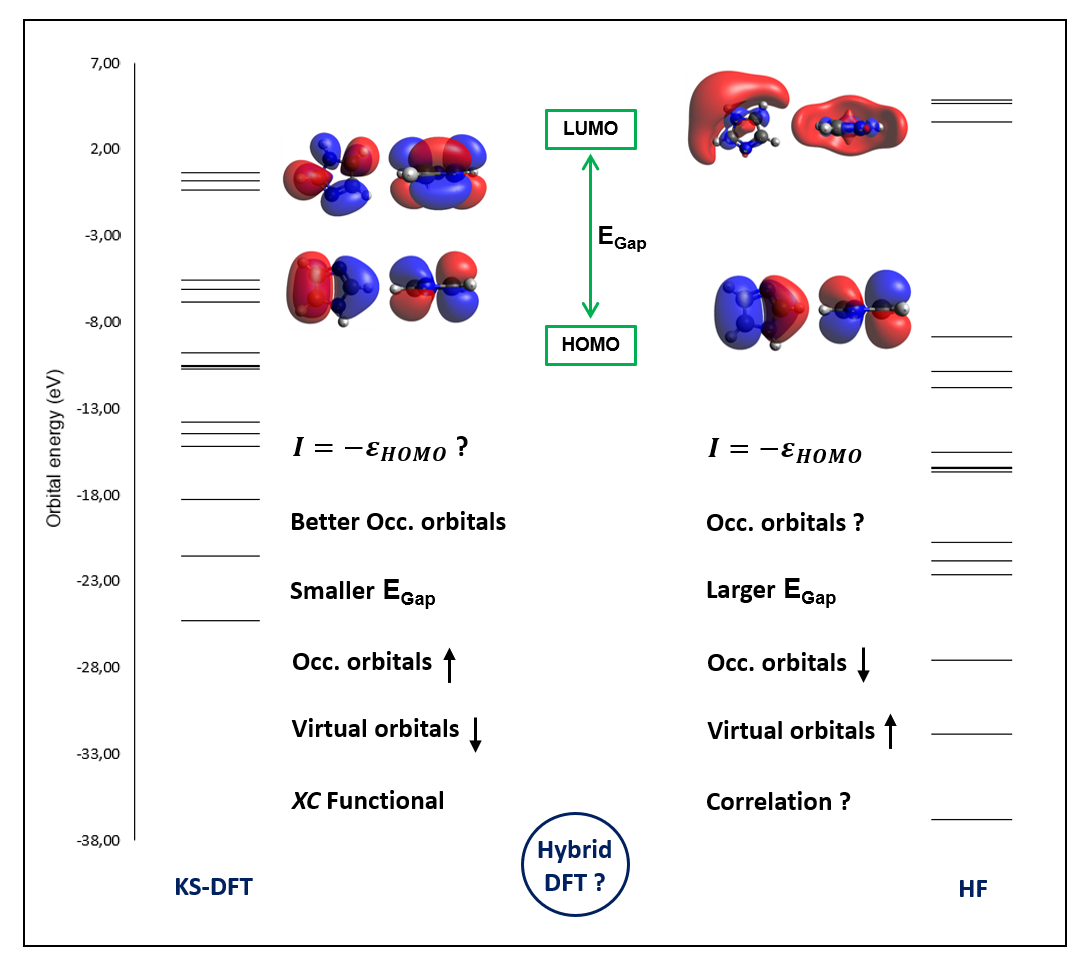
A comparative analysis on the performance of Kohn-Sham density functional theory (KS-DFT) and Hartree-Fock (HF) methods to obtain reliable energy and electronic properties has been performed in this study using a simple test case. It is crucial to re-emphasize the key differences between these methods to address common conceptual difficulties that occur among freshmen studying basic computational chemistry. The results suggested that the eigenvalue theorem in determining ionization potential could be well implemented in the HF but not in the KS-DFT method. The total energy difference between ionized and non-ionized species was an appropriate procedure to calculate the first ionization potential within the KS-DFT method. The HOMO-LUMO gap in the HF was larger than the gaps obtained from the KS-DFT method. Among all of the performed calculation methods, the B3LYP hybrid functional provided better total energy where the eigenvalues were located between the HF and the LDA/GGA functionals.
References
Jones, R. O. 2015. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 87(3), 897-923. https://doi.org/10.1103/RevModPhys.87.897
Pribram-Jones, A., Gross, D. A., & Burke, K. 2015. DFT: A theory full of holes?. Annu. Rev. Phys. Chem. 66, 283-304. https://doi.org/10.1146/annurev-physchem-040214-121420
Haunschild, R., Barth, A., & Marx, W. 2016. Evolution of DFT studies in view of a scientometric perspective. J. Cheminform. 8(52), 1-12. https://doi.org/10.1186/s13321-016-0166-y
Makkar, P., & Ghosh, N. N. 2021. A review on the use of DFT for the prediction of the properties of nanomaterials. RSC Adv. 11(45), 27897-27924. https://doi.org/10.1039/D1RA04876G
Nurrosyid, N., Fahri, M., Apriliyanto, Y.B. and Basuki, R., 2022. Novel Absorber Material Design Based on Thiazole Derivatives Using DFT/TD-DFT Calculation Methods for High-Performance Dye Sensitized Solar Cell. Indones. J. Chem. Stud. 1(1), 16-23. https://doi.org/10.55749/ijcs.v1i1.5
Apriliyanto, Y.B., Faginas-Lago, N., Evangelisti, S., Bartolomei, M., Leininger, T., Pirani, F., Pacifici, L., and Lombardi, A. 2022. Multilayer Graphtriyne Membranes for Separation and Storage of CO2: Molecular Dynamics Simulations of Post-Combustion Model Mixtures. Molecules. 27(18), 5958. https://doi.org/10.3390/molecules27185958
Faginas-Lago, N., Apriliyanto, Y.B., Lombardi, A. 2020. Carbon capture and separation from CO2/N2/H2O gaseous mixtures in bilayer graphtriyne: a molecular dynamics study. In: Gervasi, O. (ed.) ICCSA 2020. LNCS, vol. 12255, pp. 489–501. Springer, Cham. https://doi.org/10.1007/978-3-030-58820-5_36
Faginas-Lago, N., Apriliyanto, Y.B., Lombardi, A. 2021. Confinement of CO2 inside carbon nanotubes. Eur. Phys. J. D 75(5), 1–10. https://doi.org/10.1140/epjd/s10053-021-00176-7
Jensen, F. 2007. Introduction to Computational Chemistry. West Sussex: John Wiley & Sons Ltd.
Cramer C.J. 2004. Essentials of Computational Chemistry, Theories and Models, Second Edition. West Sussex: John Wiley & Sons Ltd.
Kim, J., Hong, K., Choi, S., Hwang, S. Y., & Kim, W. Y. 2015. Configuration interaction singles based on the real-space numerical grid method: Kohn–Sham versus Hartree–Fock orbitals. Phys. Chem. Chem. Phys. 17(47), 31434-31443. https://doi.org/10.1039/C5CP00352K
Chakma, U., Ali, M. H., Das, D. K., Boidya, J. R., Khan, M. B. U., Mahmud, M. S., ... & Kumer, A. 2023. First-principles calculations to investigate structural, optical and electronic properties of ZrO2, Zr0.93Si0.07O2 and Zr0.86Si0.14O2 for dye-sensitised solar cells applications. Mol. Simul. 49(15), 1389-1400. https://doi.org/10.1080/08927022.2023.2232887
Neese, F. 2018. Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 8(1), e1327. https://doi.org/10.1002/wcms.1327
Jmol: an open-source Java viewer for chemical structures in 3D. https://jmol.sourceforge.net
NIST: Atomic Spectra Database - Ionization Energies Form. https://physics.nist.gov/PhysRefData/ASD/ionEnergy.html
Downloads
Published
How to Cite
Issue
Section
License
Copyright (c) 2023 Indonesian Journal of Chemical Studies

This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.